Drug Evaluation
A guide for authors submitting to the Expert Collection
Scope
Drug Evaluations will present an evaluated overview of the clinical experience with the orphan designated compound. Although discussion should encompass basic pharmacology and pharmacokinetics, the primary focus of the review should be the clinical efficacy of the compound. For launched compounds the information should be limited to approved indications and avoid off-label discussion. More details on journal-specific scope requirement can be found at the end of this page.
Audience
The audience will consist of scientists and managers in the pharmaceutical industry and other regulatory and marketing decision makers involved in healthcare provision.
Word limit
The word limit for Drug Evaluations is 3,000-5,000 words (not including tables, figures and references).
Explore the Expert Collection
Discover all Expert Collection, journal specific guidelines.
Peer review
All articles are subject to rigorous peer review by 2-3 independent reviewers. As the Drug Evaluations include in depth analysis of a single compound, the manuscript will also be sent to the company responsible for marketing or developing the drug to check the accuracy of the data. This is for a technical check only and the final editorial decision is based entirely on the recommendation of the independent peer review. Comments remain confidential and are shared only with the corresponding author or submitting party. For a detailed description of the journal’s peer review process, authors are referred to the journal’s website.
Every article must contain
Title
Authors’ names and addresses
Structured abstract
Introduction: Authors are required to describe the significance of the topic under discussion.
Areas covered: Authors are required to describe the research discussed and the literature search undertaken.
Expert opinion: Authors are required to summarise briefly their Expert opinion section.
References must not be included in the abstract.
Keywords
Introduction
Body of the article
– Introduction to the compound: Chemistry, Pharmacodynamics, Pharmacokinetics and metabolism.
– Clinical efficacy: (Phase I studies); Phase II studies; Phase III studies; Post-marketing surveillance (depending on stage of drug and data availability).
– Safety and tolerability
– Regulatory affairs: Include information on the status of the drug, i.e., if approved, where (in which countries) it is currently approved and for what indications. This should cover Europe, US and rest of the world where appropriate.
– Conclusion: An analysis of the data presented in the review.
Expert Opinion
1. What, if any, improvement does the drug hold over other therapies?
2. In developing this drug, what are the key lessons for R&D scientists in the field? Could these lessons be applied to similar R&D in associated fields?
3. What, if any, impact is this drug/therapy likely to have on current treatment strategies?
4. How likely are physicians to prescribe the drug?
5. What data is still needed?
6. Where is the drug likely to be in 5 years’ time?
Please note that ‘opinions’ are encouraged in the Expert Opinion section, and as such, referees are asked to keep this in mind when peer reviewing the manuscript. However, authors are requested to focus their discussion on approved uses of the drug (exceptions outlined in scope section below).
Drug summary box
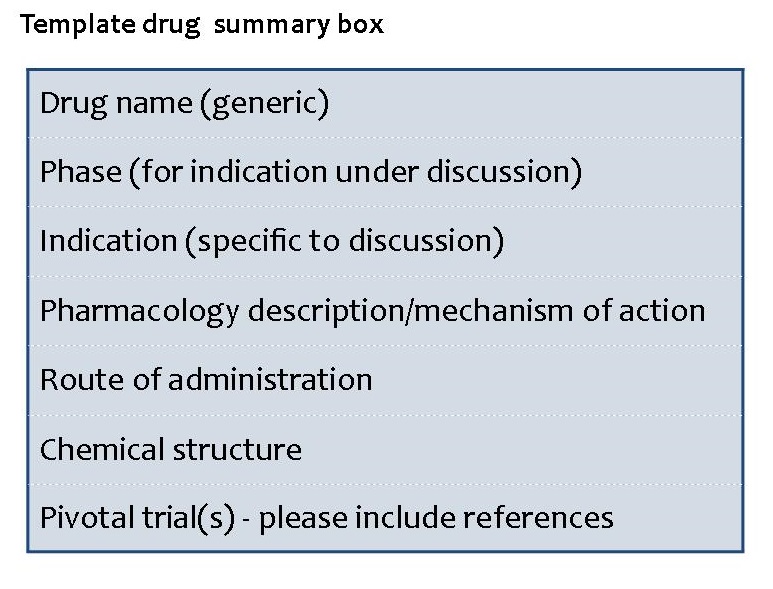
If you are unable to provide this information, editorial support will be provided.
References
Annotated bibliography
Figures and tables
Expert Opinion journal-specific scope requirements
Please note that some “Expert Opinion” journals have specific requirements for the stage which the drug has reached in the drug development pipeline. The journals will only consider submissions that describe research within this scope. Full scope details can be found on the journal’s Aims & Scope pages, but details specific to Drug Evaluations can be found below:
Expert Opinion on Investigational Drugs
Expert Opinion on Drug Metabolism & Toxicology
– Chemistry
– Pharmacodynamics (Effect of the body on the drug, including time course of absorption, bioavailability, distribution, metabolism, and excretion)
– Pharmacokinetics & metabolism: Effect of the body on the drug, including time course of absorption, bioavailability, distribution, metabolism, and excretion. To provide the reader with a visual summary of the pharmacokinetics of the drug, each paper should include a table summarising published pharmacokinetic parameters, e.g. AUC, Cmax, Cmin, Ctrough, Tmax, oral clearance CL/F, (Table 1. Summary of pharmacokinetic parameters of [drug].) ADME properties as they relate to toxicity (toxicokinetics).
– Pharmacogenetics (Effect of genetic differences on response to drugs (where data is available); clinical/population pharmacokinetics)
– Effect of genetic differences on response to drugs (where data is available).
– Clinical/population pharmacokinetics